
http://www.msg.chem.iastate.edu/GAMESS/download.html
Note in particular that GAMESS' source can be compiled for Apple systems running OS X, or for PC systems running Linux. These are not just desktop screens, but run full-fledged Unix operating systems. Therefore, they are even capable of the parallel execution of GAMESS. However, if you do not feel comfortable about compiling source code, please look here for information about precompiled binary executables for desktop systems: Macintosh
Online Services headline animator, feedBurner
Wednesday, May 28, 2008
MacMolPlt freeware
http://www.scl.ameslab.gov/~brett/MacMolPlt/
MacMolPlt is:
Below are a few samples created directly with MacMolPlt:
Simple Molecule Display with labels and displaying the bond angle:

Water with the C2v operators.


Quinone with a total electron density surface colorized with the molecular electrostatic potential value.

Check here for a list of known issues.
Click to subscribe to an email announcement list for new versions of MacMolPlt. Note this list is moderated and will only be used for announcing new releases.
Have a question or comment? Just include it in the comment section of the survey form. Make sure to include your email address if you want a reply.
If you need a version of MacMolPlt for MacOS 9 or MacOS X 10.2 and earlier you can grab the original MacMolPlt.
MacMolPlt is free, but if you use it I request that you send me a note (E-Mail or Snail mail) telling me what you like and what you don't like (What's not to like ;-).
If you use MacMolPlt for published work an appropriate reference is:
Bode, B. M. and Gordon, M. S. J. Mol. Graphics Mod., 16, 1998, 133-138.
- What is it?
- Sample Output
- New features
- Downloading MacMolPlt
- Online manual (included with the executable)
MacMolPlt is:
- A modern graphics program for plotting 3-D molecular structures and normal modes (vibrations). Modern means:
- Mouse driven interface for real-time rotation and translation.
- copy and paste functionality for interfacing to other programs such as word processors or other graphics programs (like ChemDraw).
- simple printing to color or black and white printers (publication quality).
- multiple files open at once.
- It reads a variety of file formats including any GAMESS input, log or IRC file directly to create animations of IRC's, DRC's, and optimizations. You may also import a $VEC group from any file (such as a GAMESS .DAT file). In addition xMol XYZ files, MolDen format files and Chemical Markup Language (CML) files are supported. Also some PDB file support and MDL MolFile support is included.
- Molecular point group symmetry is supported.
- You may also paste GAMESS and Gaussian-92 style cartesian coordinates directly into the program.
- Animation of Normal Modes.
- Animation of IRC's, and DRC's including orbitals.
- Simple Energy Plots (including geometrical parameters).
- Simple frequency line graph of frequency versus infrared or Raman intensity.
- Append multiple GAMESS files together to create a single animation.
- Build or modify molecules using the graphical molecule builder.
- Quickly build realistic 3D structures.
- rotate selected atoms about bonds, change bond or dihedral angles.
- translate and rotate selected subgroups.
- Build molecules from scratch using cartesian or internal coordinates
- 2D orbital, total electron density contour map display
- 3D molecular orbital, total electron density display
- density difference maps
- Molecular Electrostatic Potential Maps
- Simple GAMESS input (.inp) builder
- 3D color display with lighting and shading using OpenGL under MacOS X.
Below are a few samples created directly with MacMolPlt:
Simple Molecule Display with labels and displaying the bond angle:
Water with the C2v operators.
Quinone with a total electron density surface colorized with the molecular electrostatic potential value.
Distributions
- MacOS X:
- MacMolPlt 7.2.1 for MacOS X 10.3 and later on PowerPC or Intel processors (Universal binary) ( 5.8M )
- MacMolPlt 7.2.1 for MacOS X 10.4 and later on PowerPC or Intel processors (Universal binary) supporting quad-buffered OpenGL ( 6.9M )
- The only functional difference between these two binaries is that the latter supports hardware stereo display, but does not run on OS X 10.3.
- Linux:
- Here is a source distribution primarily targeted at Linux: wxmacmolplt 7.2.1 ( 1.5M)
- For further information on building and prebuilt binaries please refer to our Linux support page.
- Windows:
- Most users should use this Windows installer (.msi) package MacMolPlt 7.2.1 for Windows ( 4.7M)
You should be able to download it and double click it to install MacMolPlt. - If you can not run an installer on your system here is a plain zip archive. If the wxMacMolPlt.exe file fails to launch with a message saying the configuration is invalid you need to install the VS8 runtime libraries. Just run the file linked here.
- The Windows build should support quad-buffered OpenGL.
- Most users should use this Windows installer (.msi) package MacMolPlt 7.2.1 for Windows ( 4.7M)
Check here for a list of known issues.
Click to subscribe to an email announcement list for new versions of MacMolPlt. Note this list is moderated and will only be used for announcing new releases.
Have a question or comment? Just include it in the comment section of the survey form. Make sure to include your email address if you want a reply.
If you need a version of MacMolPlt for MacOS 9 or MacOS X 10.2 and earlier you can grab the original MacMolPlt.
Remember:
MacMolPlt is free, but if you use it I request that you send me a note (E-Mail or Snail mail) telling me what you like and what you don't like (What's not to like ;-).
If you use MacMolPlt for published work an appropriate reference is:
Bode, B. M. and Gordon, M. S. J. Mol. Graphics Mod., 16, 1998, 133-138.
Two-photon absorption strength
THE JOURNAL OF CHEMICAL PHYSICS 124, 204104 , 2006.
jchemphys_124_2041041
Two-photon absorption strength: A new tool for the quantification of two-photon absorption
Rémy Fortrie
Laboratoire de Chimie, UMR 5182 du CNRS, École Normale Supérieure de Lyon, 46 Allée d’Italie, F-69364 Lyon Cedex 07, France
Henry Chermettea
Université Claude Bernard Lyon 1 and UMR 5182 du CNRS Chimie Physique Théorique, Bât. Paul Dirac (210), 43 Boulevard du 11 Novembre 1918, F-69622 Villeurbanne Cedex, France
Received 21 December 2005; accepted 29 March 2006; published online 22 May 2006��
In this paper, we define the two-photon absorption strength, a new characterization tool, similar to the oscillator strength, but for two-photon absorption. It allows the quantification of the two-photon absorption properties of molecular systems which are one-photon transparent. Its definition is such that the corresponding numerical values are around 100 for small molecules. We also show that this new theoretical tool allows the direct comparison of experimental and theoretical data without requiring the introduction of any arbitrary band width. As an example, the experimental and theoretical ��AM1 + CNDO / S and HF + CIS / 3-21G�� two-photon absorption properties of the 2 , 2��-bi��9,9-dihexylfluorene�� molecule are compared. © 2006 American Institute of Physics. ��DOI: 10.1063/1.2198530�� THE JOURNAL OF CHEMICAL PHYSICS 124, 204104 ��2006��
jchemphys_124_2041041
Two-photon absorption strength: A new tool for the quantification of two-photon absorption
Rémy Fortrie
Laboratoire de Chimie, UMR 5182 du CNRS, École Normale Supérieure de Lyon, 46 Allée d’Italie, F-69364 Lyon Cedex 07, France
Henry Chermettea
Université Claude Bernard Lyon 1 and UMR 5182 du CNRS Chimie Physique Théorique, Bât. Paul Dirac (210), 43 Boulevard du 11 Novembre 1918, F-69622 Villeurbanne Cedex, France
Received 21 December 2005; accepted 29 March 2006; published online 22 May 2006��
In this paper, we define the two-photon absorption strength, a new characterization tool, similar to the oscillator strength, but for two-photon absorption. It allows the quantification of the two-photon absorption properties of molecular systems which are one-photon transparent. Its definition is such that the corresponding numerical values are around 100 for small molecules. We also show that this new theoretical tool allows the direct comparison of experimental and theoretical data without requiring the introduction of any arbitrary band width. As an example, the experimental and theoretical ��AM1 + CNDO / S and HF + CIS / 3-21G�� two-photon absorption properties of the 2 , 2��-bi��9,9-dihexylfluorene�� molecule are compared. © 2006 American Institute of Physics. ��DOI: 10.1063/1.2198530�� THE JOURNAL OF CHEMICAL PHYSICS 124, 204104 ��2006��
molcas
http://www.teokem.lu.se/molcas/
MOLCAS-7 is the latest version of the MOLCAS quantum chemistry software program. It has been released for distribution in the summer of 2007. MOLCAS-7 is a quantum chemistry code that allows studies of molecular systems using a variety of quantum chemical models ranging from SCF/DFT to coupled cluster and multiconfigurational SCF (RASSCF) with dynamical electron correlation treated with multi-reference CI or second order perturbation theory (MS-CASPT2). MOLCAS emphasizes the multiconfigurational approach for quantum chemical calculations, which allows studies of systems where a single configuration does not give a good representation of the electronic structure. Examples are excited states, transition states for chemical reactions, heavy element systems (transition metals, lanthanides, actinides), and much more.
Molcas is distributed as a source code with the set of configuration files for different hardware and compilers. In fact, Molcas is running under almost all Unix and Linux platforms, and also under MS Windows and Mac OS 10 operating systems.
MOLCAS is in particular designed to study potential surfaces for excited states.
MOLCAS-7 is the latest version of the MOLCAS quantum chemistry software program. It has been released for distribution in the summer of 2007. MOLCAS-7 is a quantum chemistry code that allows studies of molecular systems using a variety of quantum chemical models ranging from SCF/DFT to coupled cluster and multiconfigurational SCF (RASSCF) with dynamical electron correlation treated with multi-reference CI or second order perturbation theory (MS-CASPT2). MOLCAS emphasizes the multiconfigurational approach for quantum chemical calculations, which allows studies of systems where a single configuration does not give a good representation of the electronic structure. Examples are excited states, transition states for chemical reactions, heavy element systems (transition metals, lanthanides, actinides), and much more.
Molcas is distributed as a source code with the set of configuration files for different hardware and compilers. In fact, Molcas is running under almost all Unix and Linux platforms, and also under MS Windows and Mac OS 10 operating systems.
Excited States and Electronic Spectra
MOLCAS is in particular designed to study potential surfaces for excited states.
- Energies may be obtained using all the wave function methods. Geometry optimization is possible also for state average RASSCF energies.
- Transition properties are computed at the RASSCF level using the RASSCF State Interaction Method, which is unique to the MOLCAS program.
- The same code can also be used to compute spin-orbit coupling using a effective one-electron SO Hamiltonian and so called Atomic Mean Field Integrals (AMFI).
- Automatic search at the RASSCF level for energy barriers, conical intersections, etc on excited state surfaces.
- Vibrationally resolved electronic spectra may be obtained using the MULA code for computing transition dipole moments between harmonic vibrational levels of two electronic states.
lectures on Quantum mechanical methods
http://www.chem.umn.edu/groups/gao/qmmm_notes/LEC_MO.html
Quantum mechanical methods for the study of molecules can be divided into two categories: ab initio and semiempirical models. Ab initio methods refer to quantum chemical methods in which all the integrals are exactly evaluated in the course of a calculation. Ab initio methods include Hartree-Fock (HF) or molecular orbital (MO) theory, configuration interaction (CI) theory, perturbation theory (PT), and density functional theory (DFT). Ab initio methods that include correlation can have an accuracy comparable with experiment in structure and energy predictions. However, a drawback is that ab initio calculations are extremely demanding in computer resources, especially for large molecular systems. Semiempirical quantum chemical methods lie between ab initio and molecular mechanics (MM). Like MM, they use experimentally derived parameters to strive for accuracy; like ab initio methods, they are quantum-mechanical in nature. Semiempirical methods are computationally fast because many of the difficult integrals are neglected. The error introduced is compensated through the use of parameters. Thus, semiempirical procedures can often produce greater accuracy than ab initio calculations at a similar level.
1. Hartree-Fock Theory
We start with the stationary state Schrodinger equation:
 (1)
(1)
The nonrelativistic, time independent, fixed nuclei Hamiltonian is:
 (2)
(2)
where A and B designate nuclei, and I and j electrons, Z are the atomic numbers, and atomic units are used in eq (1). The many-electron wave-function Ψ is approximated as a product of one-electron functions φ(I) - the orbital approximation.
 (3)
(3)
where A is the antisymmetrizer, ensuring the wavefunction obeys the Pauli exclusion principle, and O(s) is a spin projection operator that ensures that the wavefunction remains an eigenfunction of the spin-squared operator, S2.
Each molecular orbital is then expanded as a linear combination of atom orbitals, or basis functions, χu:
 (4)
(4)
Utilizing the variational principle <Ψ| H |Ψ>/<Ψ|Ψ> ³ E(exp), Ψ is varied with respect to C, and an eigenvalue equation that yields the molecular orbitals and energies is obtained.
 (5)
(5)
where f is an effective one-electron Fock operator with matrix elements given by
 (6)
(6)
The one-electron matrix elements are give by
 (7)
(7)
and two-electron integrals by
 (8)
(8)
P is the first-order Fock-Dirac (or one-particle) density matrix
 (9)
(9)
where ni is occupation number, 2, 1, or 0.
The Fock equations can be solved by matrix diagonalization of the matrix equations
 (10)
(10)
where S is the overlap matrix and C is a square matrix with the ith column being the MO coefficients of the ith molecular orbital.
The following steps are common to all MO procedures:
1. Calculate the integrals needed to form the Fock matrix F.
2. Calculate the overlap matrix S.
3. Diagonalize S. W+SW = D
4. Form S-1/2 = WD-1/2W+.
5. Form the F matrix from equation (6).
6. Form F’ = S-1/2FS-1/2.
7. Diagonalize F’ for the MO eigenvalues E, V+F’V = E.
8. Back transform V to obtain the MO coefficients C, C = S-1/2V.
9. Form the density matrix P.
2. Approximate MO theories.
Pople and coworkers introduced a series of zero-differential overlap approximations in 1965. The hierarchy of integral approximations and the effect on the Fock matrix formation are given below.
Complete Neglect of Differential Overlap (CNDO) method.
In this approximation, all integrals involving different atomic orbitals, χu, are ignored; <uv|st> = δ(u,v) δ(s,t)<uv|st> = <uu|ss>. Thus, the overlap matrix becomes the unit matrix, S = 1, and the Fock matrix elements:
 (11a)
(11a)
 (11b)
(11b)
Parameterization and implementation scheme of the CNDO method was also proposed by Pople:
* All two-center two-electron integrals between a pair of atoms are set equal: <uv|st> (= <uu|ss>) = γAB, where γAB is a function of atoms A and B, depending on the interatomic distance RAB.
* The off-diagonal one-electron, or resonance integrals, are set proportional to S:
 (12)
(12)
where βA is a parameter that depends only on the nature of atom A.
* Electron-core attraction interactions for a given pair of atoms are set equal: <u| VB|v> = δ(u,v) VAB, with VAB = <uA| ZB/R | vA >.
* The one-center core integral Uuu is approximated by assuming that the same atomic orbital for the atom are appropriate for the positive ion: Uuu = -Iu - (ZA -1)γAA. This represents the energy required to remove an electron from atomic orbital χu in the fully ionized atom. An alternative would have been to derive Uuu from the electron affinity - the energy gained when a fully ionized atom captured an electron. CNDO/2 utilized the average of the two.
* A minimum basis set of valence orbitals was used chosen, using Slater type orbitals (STO).
 (13)
(13)
where Y(θ,φ) is the real spherical harmonics.
These approximations reduce the Fock equations from the full Roothaan form to
 (14a)
(14a)
 (14b)
(14b)
 (14c)
(14c)
However, it was soon realized that the electron-core attraction and the electron-electron repulsion are imbalanced in the CNDO ZDO approximation, and the appropriate two-center two-electron integral was used to approximate the electron-core attraction.
 (15)
(15)
The total electronic energy was calculated from
 (16)
(16)
Intermediate Neglect of Differential Overlap (INDO).
In INDO, the constraint in CNDO that monocentric two-electron integrals be equal was removed. Consequently, for atoms with s and p orbitals, there five unique two-electron one-center integrals: <ss|ss> = gss, <ss|pp> = gsp, <pp|pp> = gpp, <pp|p’p’> = gp2, p ¹ p’, and <sp|sp> = hsp. The diagonal Fock matrix element in INDO is
 (17)
(17)
in which eq (15) was used in place of the electron-core attraction integral.
 (18)
(18)
Since INDO and CNDO execute on a computer at about the same speeds and INDO contains some important integrals neglected in the CNDO method, INDO performs much better than CNDO especially in prediction of molecular spectral properties. One most successful program is that developed by Ridley and Zerner in 1973. The model, CNDO/S, was parameterized by carrying out CI-singles (CIS) calculations, and based one atomic spectroscopic data. In general, the INDO/S model reproduces the excitation energies of transitions below 40,000 cm-1 within 2000 cm-1. Another well-known program at this level is Dewar’s MINDO/3 model (1975). Here, all quantities that entered the Fock matrix and the energy expression were treated as free parameters. As a result, the MINDO/3 model achieved an impressive predictive power.
Neglect of Diatomic Differential Overlap (NDDO).
The NDDO model can be derived from the replacement:
 (19)
(19)
This leads to the following Fock matrix elements:
 (20a)
(20a)
 (20b)
(20b)
 (20c)
(20c)
Here, all two-electron two-center integrals involving charge clouds arising from pairs of orbitals on an atom were retained. This model allowed lone-pair lone-pair repulsions to be represented. Since there are four valence orbitals (for an sp atom), there are 10 unique pairs, giving rise to 100 integrals. For (spd) basis, 2025 two-electron integrals are needed compared to INDO’s 4.
The NDDO model is the only model so far that really relates to an actual basis set. The first practical NDDO model was introduced by Dewar and Thiel in 1977. Thiel has continued recently to expand the original MNDO model to include d orbitals. The model was parameterized on molecular geometries, heats of formation, dipole moments, and ionization potentials. A feature in the MNDO model is that the core-core repulsion term was made a function of the electron-electron repulsion integrals:
 (21)
(21)
A major fatal problem in the MNDO model is its inability to reproduce hydrogen bonding interactions due to a spurious repulsion at just outside chemical bonding distances. The solution to this problem was to assign a number of spherical Gaussians to mimic the correlation effects. This is the Austin Model 1 (AM1):
 (22)
(22)
This increased the number of parameters from the original 7 to 13-16 per atom. The AM1 model is undoubtedly a much improvement over the MNDO model for a wide range of properties. Stewart’s Parametric Method Number 3 (PM3) model treat all quantities that enter the Fock matrix as free parameters, while MNDO and AM1 derive the one-center two-electron integrals from atomic spectroscopy.
3. Integral evaluation
While in CNDO and INDO, most electron repulsion integrals were neglected, and those that are left had a common value (γAB) for a given pair of atoms. On the other hand, in NDDO, there are 22 non-vanishing integrals between first-row atoms. The NDDO electron repulsion integrals are determined in semiempirical programs in terms of multipole-multipole interactions, a method introduced by Dewar and Thiel in 1977. The atomic orbitals used in this treatment are the Slater-Zener orbitals, which are products of a radial function Rnl(r) and a normalized real spherical harmonic Ylm(θ,φ), with quantum numbers n, l, and m.
 (22)
(22)


where ξ is the orbital exponent of the Slater AO, and Pl|m|(cos θ) is an associated Legendre function. Within MNDO, AM1, and PM3, only s and p basis AO were used (l = 0, 1). The multipole moments Mlm of a charge distribution ρ(r, θ, φ) is defined by
 (23)
(23)
The notation of multipole moments may be compared with the common notations: M00 = q;
M10 = μz; M11 = μx; M1-1 = μy; etc.
The semiempirical formalism for the NDDO repulsion integrals were derived by first assuming that the two interacting charge distributions do not overlap. The solution to this problem is to introduce normalized real spherical harmonics and a bipolar expansion for 1/r12.
After substituting the 1/r12 expansion into the two-electron integral expression and comparison with the definition of multipoles, the two electron repulsion integrals can be expressed by
 (24)
(24)
where the coefficients depends on l, m and dlm. Eq (24) has the correct asymptotic behavior for RAB ® ¥. For short distances, the assumption of zero diatomic overlap is no longer valid. Therefore, eq (23) must be modified semiempirically to ensure the behavior at short distances. The modification was provided by applying the Klopman formula for monopole-monopole interactions between charge distributions uu and vv:
 (25)
(25)
The nonvanishing multipoles Mlm in the NDDO integrals are represented by configurations of 2l point charges of magnitude e/2l. The interactions between the multipoles are calculated by applying the Klopman formula to each point charge pair.
 (25)
(25)
There are two values needed to be determined in this treatment, the distance separating the point charges for each multipole configuration and the parameters ρ0. The first was determined analytically by comparing the exact expression for the multipoles defined by eq (23), and the results from the point charges. These distances depend on the orbital exponents. ρ0 were chosen to yield the correct one-center limit for the monopole-monopole, dipole-dipole, and quadrupole-quadrupole interactions.
Consider, for example, the integral <spz|spz>. The only nonvanishing multipole for a charge distribution of sp is the dipole term μz. Therefore, <spz|spz> = [ μz, μz ].
In hybrid QM/MM application, the one-electron integral between a charge distribution uv in the QM region and a point charge qmm in the MM region is approximated, consistent with the MNDO/AM1/PM3 procedure, by the two-electron integrals: <u|qmm/Rmm|v> = <uv|smmsmm>. The relevant integrals, thus, include <ss|ss>, <pπ pπ|ss>, <pzpz|ss>, and <spz|ss> types. In ab initio/DFT hybrid QM/MM models, the one-electron integrals are, of course, computed analytically as do all other integrals (Freindorf, J. Comput. Chem. 17, 386 (1996)).
 (26)
(26)
4. Computer programs.
The MOPAC package developed by J. J. P. Stewart was a public domain program up to MOPAC 7, and MOPAC 93. It contains the MINDO/3, MNDO, AM1, and PM3 models, which allows a variety of properties to be computed. There is also a commercial version of MOPAC as well as AMPAC. The latter contains the newest SAM1 model. The most familiar ab initio package is the Gaussian series of programs. More than a dozen of other similar programs are available. A public domain ab initio program, the GAMESS program, is also available. For spectroscopy calculations, Zerner’s INDO/S would be an excellent choice. The most sophisticated ab initio package on spectroscopy would be the MOLCAS from Roos.
III. Semiempirical Molecular Orbital Methods
Lecture Notes
Quantum mechanical methods for the study of molecules can be divided into two categories: ab initio and semiempirical models. Ab initio methods refer to quantum chemical methods in which all the integrals are exactly evaluated in the course of a calculation. Ab initio methods include Hartree-Fock (HF) or molecular orbital (MO) theory, configuration interaction (CI) theory, perturbation theory (PT), and density functional theory (DFT). Ab initio methods that include correlation can have an accuracy comparable with experiment in structure and energy predictions. However, a drawback is that ab initio calculations are extremely demanding in computer resources, especially for large molecular systems. Semiempirical quantum chemical methods lie between ab initio and molecular mechanics (MM). Like MM, they use experimentally derived parameters to strive for accuracy; like ab initio methods, they are quantum-mechanical in nature. Semiempirical methods are computationally fast because many of the difficult integrals are neglected. The error introduced is compensated through the use of parameters. Thus, semiempirical procedures can often produce greater accuracy than ab initio calculations at a similar level.
1. Hartree-Fock Theory
We start with the stationary state Schrodinger equation:
The nonrelativistic, time independent, fixed nuclei Hamiltonian is:
where A and B designate nuclei, and I and j electrons, Z are the atomic numbers, and atomic units are used in eq (1). The many-electron wave-function Ψ is approximated as a product of one-electron functions φ(I) - the orbital approximation.
where A is the antisymmetrizer, ensuring the wavefunction obeys the Pauli exclusion principle, and O(s) is a spin projection operator that ensures that the wavefunction remains an eigenfunction of the spin-squared operator, S2.
Each molecular orbital is then expanded as a linear combination of atom orbitals, or basis functions, χu:
Utilizing the variational principle <Ψ| H |Ψ>/<Ψ|Ψ> ³ E(exp), Ψ is varied with respect to C, and an eigenvalue equation that yields the molecular orbitals and energies is obtained.
where f is an effective one-electron Fock operator with matrix elements given by
The one-electron matrix elements are give by
(7)and two-electron integrals by
P is the first-order Fock-Dirac (or one-particle) density matrix
where ni is occupation number, 2, 1, or 0.
The Fock equations can be solved by matrix diagonalization of the matrix equations
where S is the overlap matrix and C is a square matrix with the ith column being the MO coefficients of the ith molecular orbital.
The following steps are common to all MO procedures:
1. Calculate the integrals needed to form the Fock matrix F.
2. Calculate the overlap matrix S.
3. Diagonalize S. W+SW = D
4. Form S-1/2 = WD-1/2W+.
5. Form the F matrix from equation (6).
6. Form F’ = S-1/2FS-1/2.
7. Diagonalize F’ for the MO eigenvalues E, V+F’V = E.
8. Back transform V to obtain the MO coefficients C, C = S-1/2V.
9. Form the density matrix P.
- Check P for convergence. If not converged, repeat steps 5-10 until self-consistent electronic field (SCF) is obtained.
2. Approximate MO theories.
Pople and coworkers introduced a series of zero-differential overlap approximations in 1965. The hierarchy of integral approximations and the effect on the Fock matrix formation are given below.
Complete Neglect of Differential Overlap (CNDO) method.
In this approximation, all integrals involving different atomic orbitals, χu, are ignored; <uv|st> = δ(u,v) δ(s,t)<uv|st> = <uu|ss>. Thus, the overlap matrix becomes the unit matrix, S = 1, and the Fock matrix elements:
(11a)Parameterization and implementation scheme of the CNDO method was also proposed by Pople:
* All two-center two-electron integrals between a pair of atoms are set equal: <uv|st> (= <uu|ss>) = γAB, where γAB is a function of atoms A and B, depending on the interatomic distance RAB.
* The off-diagonal one-electron, or resonance integrals, are set proportional to S:
where βA is a parameter that depends only on the nature of atom A.
* Electron-core attraction interactions for a given pair of atoms are set equal: <u| VB|v> = δ(u,v) VAB, with VAB = <uA| ZB/R | vA >.
* The one-center core integral Uuu is approximated by assuming that the same atomic orbital for the atom are appropriate for the positive ion: Uuu = -Iu - (ZA -1)γAA. This represents the energy required to remove an electron from atomic orbital χu in the fully ionized atom. An alternative would have been to derive Uuu from the electron affinity - the energy gained when a fully ionized atom captured an electron. CNDO/2 utilized the average of the two.
* A minimum basis set of valence orbitals was used chosen, using Slater type orbitals (STO).
where Y(θ,φ) is the real spherical harmonics.
These approximations reduce the Fock equations from the full Roothaan form to
However, it was soon realized that the electron-core attraction and the electron-electron repulsion are imbalanced in the CNDO ZDO approximation, and the appropriate two-center two-electron integral was used to approximate the electron-core attraction.
The total electronic energy was calculated from
Intermediate Neglect of Differential Overlap (INDO).
In INDO, the constraint in CNDO that monocentric two-electron integrals be equal was removed. Consequently, for atoms with s and p orbitals, there five unique two-electron one-center integrals: <ss|ss> = gss, <ss|pp> = gsp, <pp|pp> = gpp, <pp|p’p’> = gp2, p ¹ p’, and <sp|sp> = hsp. The diagonal Fock matrix element in INDO is
(17)in which eq (15) was used in place of the electron-core attraction integral.
Since INDO and CNDO execute on a computer at about the same speeds and INDO contains some important integrals neglected in the CNDO method, INDO performs much better than CNDO especially in prediction of molecular spectral properties. One most successful program is that developed by Ridley and Zerner in 1973. The model, CNDO/S, was parameterized by carrying out CI-singles (CIS) calculations, and based one atomic spectroscopic data. In general, the INDO/S model reproduces the excitation energies of transitions below 40,000 cm-1 within 2000 cm-1. Another well-known program at this level is Dewar’s MINDO/3 model (1975). Here, all quantities that entered the Fock matrix and the energy expression were treated as free parameters. As a result, the MINDO/3 model achieved an impressive predictive power.
Neglect of Diatomic Differential Overlap (NDDO).
The NDDO model can be derived from the replacement:
This leads to the following Fock matrix elements:
(20a) (20b) (20c)Here, all two-electron two-center integrals involving charge clouds arising from pairs of orbitals on an atom were retained. This model allowed lone-pair lone-pair repulsions to be represented. Since there are four valence orbitals (for an sp atom), there are 10 unique pairs, giving rise to 100 integrals. For (spd) basis, 2025 two-electron integrals are needed compared to INDO’s 4.
The NDDO model is the only model so far that really relates to an actual basis set. The first practical NDDO model was introduced by Dewar and Thiel in 1977. Thiel has continued recently to expand the original MNDO model to include d orbitals. The model was parameterized on molecular geometries, heats of formation, dipole moments, and ionization potentials. A feature in the MNDO model is that the core-core repulsion term was made a function of the electron-electron repulsion integrals:
A major fatal problem in the MNDO model is its inability to reproduce hydrogen bonding interactions due to a spurious repulsion at just outside chemical bonding distances. The solution to this problem was to assign a number of spherical Gaussians to mimic the correlation effects. This is the Austin Model 1 (AM1):
(22)This increased the number of parameters from the original 7 to 13-16 per atom. The AM1 model is undoubtedly a much improvement over the MNDO model for a wide range of properties. Stewart’s Parametric Method Number 3 (PM3) model treat all quantities that enter the Fock matrix as free parameters, while MNDO and AM1 derive the one-center two-electron integrals from atomic spectroscopy.
3. Integral evaluation
While in CNDO and INDO, most electron repulsion integrals were neglected, and those that are left had a common value (γAB) for a given pair of atoms. On the other hand, in NDDO, there are 22 non-vanishing integrals between first-row atoms. The NDDO electron repulsion integrals are determined in semiempirical programs in terms of multipole-multipole interactions, a method introduced by Dewar and Thiel in 1977. The atomic orbitals used in this treatment are the Slater-Zener orbitals, which are products of a radial function Rnl(r) and a normalized real spherical harmonic Ylm(θ,φ), with quantum numbers n, l, and m.
where ξ is the orbital exponent of the Slater AO, and Pl|m|(cos θ) is an associated Legendre function. Within MNDO, AM1, and PM3, only s and p basis AO were used (l = 0, 1). The multipole moments Mlm of a charge distribution ρ(r, θ, φ) is defined by
The notation of multipole moments may be compared with the common notations: M00 = q;
M10 = μz; M11 = μx; M1-1 = μy; etc.
The semiempirical formalism for the NDDO repulsion integrals were derived by first assuming that the two interacting charge distributions do not overlap. The solution to this problem is to introduce normalized real spherical harmonics and a bipolar expansion for 1/r12.
After substituting the 1/r12 expansion into the two-electron integral expression and comparison with the definition of multipoles, the two electron repulsion integrals can be expressed by
(24)where the coefficients depends on l, m and dlm. Eq (24) has the correct asymptotic behavior for RAB ® ¥. For short distances, the assumption of zero diatomic overlap is no longer valid. Therefore, eq (23) must be modified semiempirically to ensure the behavior at short distances. The modification was provided by applying the Klopman formula for monopole-monopole interactions between charge distributions uu and vv:
The nonvanishing multipoles Mlm in the NDDO integrals are represented by configurations of 2l point charges of magnitude e/2l. The interactions between the multipoles are calculated by applying the Klopman formula to each point charge pair.
(25)There are two values needed to be determined in this treatment, the distance separating the point charges for each multipole configuration and the parameters ρ0. The first was determined analytically by comparing the exact expression for the multipoles defined by eq (23), and the results from the point charges. These distances depend on the orbital exponents. ρ0 were chosen to yield the correct one-center limit for the monopole-monopole, dipole-dipole, and quadrupole-quadrupole interactions.
Consider, for example, the integral <spz|spz>. The only nonvanishing multipole for a charge distribution of sp is the dipole term μz. Therefore, <spz|spz> = [ μz, μz ].
In hybrid QM/MM application, the one-electron integral between a charge distribution uv in the QM region and a point charge qmm in the MM region is approximated, consistent with the MNDO/AM1/PM3 procedure, by the two-electron integrals: <u|qmm/Rmm|v> = <uv|smmsmm>. The relevant integrals, thus, include <ss|ss>, <pπ pπ|ss>, <pzpz|ss>, and <spz|ss> types. In ab initio/DFT hybrid QM/MM models, the one-electron integrals are, of course, computed analytically as do all other integrals (Freindorf, J. Comput. Chem. 17, 386 (1996)).
(26)4. Computer programs.
The MOPAC package developed by J. J. P. Stewart was a public domain program up to MOPAC 7, and MOPAC 93. It contains the MINDO/3, MNDO, AM1, and PM3 models, which allows a variety of properties to be computed. There is also a commercial version of MOPAC as well as AMPAC. The latter contains the newest SAM1 model. The most familiar ab initio package is the Gaussian series of programs. More than a dozen of other similar programs are available. A public domain ab initio program, the GAMESS program, is also available. For spectroscopy calculations, Zerner’s INDO/S would be an excellent choice. The most sophisticated ab initio package on spectroscopy would be the MOLCAS from Roos.
Pople CNDO INDO
Sir John Anthony Pople, FRS, (October 31, 1925 – March 15, 2004) was a theoretical chemist. Born in Burnham on Sea, Somerset, England, he attended Bristol Grammar School. He won a scholarship to Trinity College, Cambridge in 1943. He received his B. A. in 1946. Between 1945 and 1947 he work at the Bristol Aeroplane Company. He then returned to Cambridge University and was awarded his doctorate degree in mathematics in 1951. He moved to the United States of America in 1964, where he lived the rest of his life, though he retained British citizenship. Pople considered himself more of a mathematician than a chemist, but theoretical chemists consider him one of the most important of their number.
He received the Nobel Prize in Chemistry in 1998.[11]
His early paper[3] on the statistical mechanics of water, according to Michael J. Frisch, "remained the standard for many years.[2] This was his thesis topic for his Ph D at Cambridge supervised by John Lennard-Jones.[1]
He pioneered the development of more sophisticated computational methods, called ab initio quantum chemistry methods, that use basis sets of either Slater type orbitals or Gaussian orbitals to model the wave function. While in the early days these calculations were extremely expensive to perform, the advent of high speed microprocessors has made them much more feasible today. He was instrumental in the development of one of the most widely used computational chemistry packages, the "GAUSSIAN"(tm) suite of programs, including coauthorship of the first version, Gaussian 70.[7] One of his most important original contributions is the concept of a model chemistry whereby a method is rigorous evaluated across a range of molecules.[2] [8] He instigated the quantum chemistry composite methods such as Gaussian-1 (G1) and Gaussian-2 (G2). He was a founder of the Q-Chem computational chemistry program.[9]
ZINDO is a semi-empirical quantum chemistry method used in computational chemistry. It is a development of the INDO method. It stands for Zerner's Intermediate Neglect of Differential Overlap, as it was developed by Michael Zerner. [1] Unlike INDO which was really restricted to organic molecules and those containing the atoms B to F, ZINDO covers a wide range of the periodic table, even including the rare earth elements. There are two distinct versions of the method:-
The original program from the Zerner group is not widely available but the method is implemented in HyperChem and, in part, in Gaussian.
A major complaint against ZINDO in particular is that while it could reproduce the low lying spectra of larger polyenes and some organo-metallic compounds, it lacked a consistent parameterization. To obtain good results, it had been frequently necessary to fit the parameters to a given molecule, thereby reducing its generality and predictive capacity. In contrast, ab initio packages, such as GAUSSIAN, gained popularity because they would always produce the same results for a given input, even if the results were sometimes inaccurate.
He received the Nobel Prize in Chemistry in 1998.[11]
Statistical mechanics of water
His early paper[3] on the statistical mechanics of water, according to Michael J. Frisch, "remained the standard for many years.[2] This was his thesis topic for his Ph D at Cambridge supervised by John Lennard-Jones.[1]
Ab Initio Electronic Structure Theory
He pioneered the development of more sophisticated computational methods, called ab initio quantum chemistry methods, that use basis sets of either Slater type orbitals or Gaussian orbitals to model the wave function. While in the early days these calculations were extremely expensive to perform, the advent of high speed microprocessors has made them much more feasible today. He was instrumental in the development of one of the most widely used computational chemistry packages, the "GAUSSIAN"(tm) suite of programs, including coauthorship of the first version, Gaussian 70.[7] One of his most important original contributions is the concept of a model chemistry whereby a method is rigorous evaluated across a range of molecules.[2] [8] He instigated the quantum chemistry composite methods such as Gaussian-1 (G1) and Gaussian-2 (G2). He was a founder of the Q-Chem computational chemistry program.[9]
ZINDO is a semi-empirical quantum chemistry method used in computational chemistry. It is a development of the INDO method. It stands for Zerner's Intermediate Neglect of Differential Overlap, as it was developed by Michael Zerner. [1] Unlike INDO which was really restricted to organic molecules and those containing the atoms B to F, ZINDO covers a wide range of the periodic table, even including the rare earth elements. There are two distinct versions of the method:-
- ZINDO/1 - for calculating ground state properties such as bond lengths and bond angles.
- ZINDO/S (sometimes just called INDO/S) - for calculating excited states and hence electronic spectra.
The original program from the Zerner group is not widely available but the method is implemented in HyperChem and, in part, in Gaussian.
A major complaint against ZINDO in particular is that while it could reproduce the low lying spectra of larger polyenes and some organo-metallic compounds, it lacked a consistent parameterization. To obtain good results, it had been frequently necessary to fit the parameters to a given molecule, thereby reducing its generality and predictive capacity. In contrast, ab initio packages, such as GAUSSIAN, gained popularity because they would always produce the same results for a given input, even if the results were sometimes inaccurate.
CNDO/S-CI computation of UV/Vis spectra
| PM3 geometry optimization and CNDO/S-CI computation of UV/Vis spectra of large organic structures: Program description and application to poly(triacetylene) hexamer and taxotere |
| Harold Baumann *, Rainer E. Martin, François Diederich |
| Laboratorium für Organische Chemie der Eidgenössischen Technischen Hochschule, Universitätsstrasse 16, CH-8092 Zürich, Switzerland |
| email: Harold Baumann (baumann@org.chem.ethz.ch) |
*Correspondence to Harold Baumann, Laboratorium für Organische Chemie der Eidgenössischen Technischen Hochschule, Universitätsstrasse 16, CH-8092 Zürich, Switzerland
setDOI("ADOI=10.1002/(SICI)1096-987X(199903)20:4<396::AID-JCC2>3.0.CO;2-9")
| Keywords |
| poly(triacetylene); taxotere; UV/Vis spectrum; UHF-PM3; CNDO/S-CI; spin density wave |
| Abstract |
SIXW.C, a new C version of the computer program CNDUV99 (based on CNDO/S-CI with inclusion of doubly excited configurations) is described and shown to be useful for the computation of the UV/Vis spectra of fairly large molecules. The geometries of the molecules were obtained by our C version of program PM3. To demonstrate the broad applicability of program SIXW.C we have chosen two representative examples: first, a linearly conjugated oligomer of defined length; and, second, a natural product that currently plays a very significant role in cancer therapy. The first molecule is a recently synthesized linearly conjugated monodisperse hexamer of  4.6 nm in length (C126H222O12Si14), with poly(triacetylene) backbone. Despite the 36 conjugated C-atoms of the framework, it exhibits remarkable thermal and environmental stability, which allows in-depth investigation of its physical properties. Therefore, this made it a very attractive candidate for comparison of the theoretically calculated and experimentally measured UV/Vis spectra, as there is still considerable interest in predicting linear optical properties of large conjugated organic molecules at the border to polymers. The agreement between experimental and calculated spectra is better with the UHF-PM3- rather than with the RHF-PM3-optimized structure of the molecule, showing, for the former, excellent agreement between the experimental and theoretical longest wavelength maximum ( 4.6 nm in length (C126H222O12Si14), with poly(triacetylene) backbone. Despite the 36 conjugated C-atoms of the framework, it exhibits remarkable thermal and environmental stability, which allows in-depth investigation of its physical properties. Therefore, this made it a very attractive candidate for comparison of the theoretically calculated and experimentally measured UV/Vis spectra, as there is still considerable interest in predicting linear optical properties of large conjugated organic molecules at the border to polymers. The agreement between experimental and calculated spectra is better with the UHF-PM3- rather than with the RHF-PM3-optimized structure of the molecule, showing, for the former, excellent agreement between the experimental and theoretical longest wavelength maximum ( max), which differ by only 16 nm (802 cm-1). This result is of fundamental interest, because the electronic structure reveals a spin-polarized character, usually referred to as spin density wave. In addition to the flat poly(triacetylene) oligomer, we have calculated N-tert-butoxycarbonyl-10-deacetyl-N-debenzoyltaxol (C43H53NO14, taxotere) as an example for a three-dimensional molecule for which, until now, only MM2 force-field computations were performed. It is closely related to the potent inhibitor of cell division, taxol, which was isolated first in 1971 from the stem bark of the yew Taxus brevifolia Nutt. Taxotere is one of the key weapons in modern cancer therapy. ©1999 John Wiley & Sons, Inc. J Comput Chem 20: 396-411, 1999 max), which differ by only 16 nm (802 cm-1). This result is of fundamental interest, because the electronic structure reveals a spin-polarized character, usually referred to as spin density wave. In addition to the flat poly(triacetylene) oligomer, we have calculated N-tert-butoxycarbonyl-10-deacetyl-N-debenzoyltaxol (C43H53NO14, taxotere) as an example for a three-dimensional molecule for which, until now, only MM2 force-field computations were performed. It is closely related to the potent inhibitor of cell division, taxol, which was isolated first in 1971 from the stem bark of the yew Taxus brevifolia Nutt. Taxotere is one of the key weapons in modern cancer therapy. ©1999 John Wiley & Sons, Inc. J Comput Chem 20: 396-411, 1999 |
Received: 17 December 1997; Revised: 29 September 1998; Accepted: 1 October 1998
| Digital Object Identifier (DOI) |
10.1002/(SICI)1096-987X(199903)20:4<396::AID-JCC2>3.0.CO;2-9 About DOI
Alphabetical list of 3D data formats
Alphabetical list of 3D API specifications and data formats
http://local.wasp.uwa.edu.au/~pbourke/dataformats/
http://local.wasp.uwa.edu.au/~pbourke/dataformats/
| 3D2 | Stereo CAD-3D object format |
| 3DML | Flatland 3DML language |
| AC3D | AC3D |
| 3DS ASE ASC | 3D-Studio File Format 3D Studio Max Ascii Export Format 3D Studio Ascii Format |
| ALC | Alchemy III molecule file format |
| AL2 | Alchemy 2000 molecule file format |
| BMF | BMF by David Farrell, exported from 3dStudio by "view3ds'. |
| CDF | Cyberspace Description Format |
| CGM | Computer Graphics Metafile (ISO/IEC standardfor vector graphics) |
| cinema4d | Cinema4d file format from "Maxon Computer" |
| COB | Calgari trueSpace2 File Format |
| DAE | Sony / Khronos Collada |
| cube | Gaussian cube file format for volumetric data |
| DF3 | Povray DF3 density (volumetric) format |
| Direct-X | Microsofts answer to QuickDraw3D and OpenGL |
| DMO | Duke Nukem 3D or Redneck Rampage |
| DWF | Format used by AutoDesks attempt at an internet format for it's models, used by the WHIP viewer. |
| DXF2000 Release 14 Release 12 Release 10 | DXF, AutoDesk/AutoCAD interchange format in the various format versions that have appeared over the years. |
| Minimal 3D DXF | The minimal requirements to represent 3D geometry in DXF, useful for creating geometry for commercial packages from your own software. |
| EGDR | MOLA Experiment Gridded Data Record |
| FACT | From ElectricImage |
| FBX | Autodesk FBX |
| FFIVW | File Format for the Interchange of Virtual Worlds |
| fld | AVS Field format. |
| FLT | OpenFlight format by MulitGen Inc |
| GEO | Videoscape geo an early Amiga 3D animation program written by Allen Hastings. |
| Geom | Geom format as used by "Stereo", an OpenGL interactive stereo pair package. |
| GLF | 3D font format for the GLF library |
| GOCAD | GOCAD ascii data format |
| HIV | HyperChem molecular format |
| HPGL | Hewlett Packard Graphics Language for platters |
| hf | Hyperfun: Language for F-rep Geometric Modelling |
| IGES | Initial Graphics Exchange Specification by the National Bureau of Standards |
| ILDA | International Laser Display Association |
| Infini3D | Infini3D internal format |
| Inventor | Inventor ASCII format from SGI |
| IRIT | IRIT interchange format by Gershon Elber |
| LWOB Lightwave 5.x | LightWave Object File format Importing geometry into Lightwave 5.x |
| MDL | From Cornell University and Indiana University |
| MGF | Materials and Geometry format originally appearing as part of the Radiance package. By Greg Ward, Lawrence Berkeley Laboratory. |
| MI | Mental Images, as used by Mental Ray, SoftImage, and others |
| MOL2 | Tripos mol2 molecule format |
| MovieBYU | Format from Birmingham Young University to represent polygons, originally designed for FORTRAN file IO. |
| MS3D | MilkShape 3D format |
| MSLD | Manchester Scene Description Language |
| MTL | Lightwave / OBJ material file |
| NDO | NENDO by Izware |
| nff | For Eric Haines' SPD package |
| nff | Neutral ASCII File Format |
| enff | Extensions to Neutral File Format |
| NUAGES | Format for the NUAGES software, a tool for 3D reconstruction from parallel cross-sections |
| OBJ | Wavefront .obj file format specification for the Advanced Visualizer software. |
| OFF | OFF format as used by the Geometry Center |
| OFF | OFF format specification originally developed by WSE. |
| OOGL | As used by GeomView |
| PEDR | MOLA Precision Experiment Data Records from NASA |
| PDB (V2.1) | Protein Data Base (Atomic Coordinate File) |
| PHD | PolyHedra Database (NetLib) |
| PI | Format for the polyray raytracer by Alexander Enzmann |
| PLG, FIG, WLD | Virtual World formats as used in Gossamer, Rend386, and others. |
| PLY | Polygon File Format also known as the Stanford Triangle Format. |
| Poly | Another Polygon Format from the University of Iowa, Image Analysis Facility |
| PS | Introduction to Postscript |
| POVRAY (v3.6) | Scene format for the Persistence Of Vision RAYtracer (and derivatves). |
| PVL | Processed VoLume (and other RAW formats) as used by the Drishti volume rendering software. |
| Macperspective | Translator for this undocumented format. |
| PowerFlip | Data format (SGI) |
| PRT | PRT raytracer format by Kory Hamzeh |
| PRT | Unigraphics "parts" file format. |
| q3o | Quick3D Object File and Scene file format (.q3s) |
| QuickDraw3D Primitive summary | Apple's Quickdraw 3D meta format |
| radio | Radio format by Anthony D'Agostino |
| RAD V3.1 RAD V2.5 ArchiCAD -> Radiance StrataStudio -> Radiance | Radiance scene description by Greg Ward. ArchiCAD to Radiance converter StrataStudio to Radiance converter |
| RAW | PovRay raw triangle format |
| RAY RAY summary | RayShade scene description format, a solid modelling by Craig Kolb. |
| RIB | Pixar RenderMan scene description (RenderMan Interface Bytestream) |
| Rotater | Macintosh interactive line and point viewer by Craig Kloeden. |
| rsd | Playstation |
| SAT | ACIS 3D format for viewing and transferring solid information |
| SCENE | A proposed format for 3D geometry |
| SCN | SCeNe format designed to replace SFF for the Rtrace ray-tracer. |
| SDML Old SDML | Spatial Data Modelling Language |
| SHP | ERSI Shapefile |
| SLC | SLiCe format |
| STL | Industry standard format for stereoLithography. |
| STP | SteinLib format |
| STEP | Standard for exchange of product model |
| Super3D | Text export format used by the Macintosh modeller Super3D. |
| SURF | Export format from 3D-XplorMath |
| Tachyon | Preliminary scene format for tachyon by John E. Stone |
| FORM TDDD | By Impulse's Turbo Silver for Sense8's WorldToolKit Neutral File Format specification |
| tet | Format for tetrahedra, originating at the Computer Science department of Williams College. |
| TIN | Triangular Irregular Network |
| TM | LONI triangle surface model format to represent surface models. |
| TP | TecPlot file format |
| TRI | Triangle format |
| U3D | Universal 3D |
| UNREAL | UNREAL File Format |
| V | VIVID file format by Stephen Coy |
| VEF | Vertex - Edge - Face format |
| Vision3D | Text format for the Macintosh Vision3D modeller |
| VLA | Digistar II VLA format |
| vmd | VMD - WinOSi (XOSi, MacOSi) raytacer by Michael Granz. |
| vol | Paul Bourke volumetric data format |
| VRI | Virtual Reality Interchange Language |
| WLD | Morfit's WorldBuilder format |
| WRM | World Reference Model by Multigen Inc. |
| VRML V1.0 VRML 97 | The Virtual Reality Modelling Language |
| WMF | Windows Metafile Format |
| x3d | WEB3D consortium |
| XYZ | XYZ molecular format |
| YASRT | YASRT - Yet Another Simple Ray Tracer |
| YAODL | SGI PowerFlip format |
QuteMol
QuteMol is an open source (GPL), interactive, high quality molecular visualization system. QuteMol exploits the current GPU capabilites through OpenGL shaders to offers an array of innovative visual effects. QuteMol visualization techniques are aimed at improving clarity and an easier understanding of the 3D shape and structure of large molecules or complex proteins.
- Real Time Ambient Occlusion
- Depth Aware Silhouette Enhancement
- Ball and Sticks, Space-Fill and Liquorice visualization modes
- High resolution antialiased snapshots for creating publication quality renderings
- Automatic generation of animated gifs of rotating molecules for web pages animations
- Real-time rendering of large molecules and protein (>100k atoms)
- Standard PDB input
- Quick installers for Win and Mac OS X (intel) (new!)
- Support as a plugins of the NanoEngineer-1 the modeling and simulation program for nano-composites (new!)
FSL library of analysis tools for FMRI, MRI and DTI brain imaging data.
http://www.fmrib.ox.ac.uk/fsl/index.html
Introduction
FSL is a comprehensive library of analysis tools for FMRI, MRI and DTI brain imaging data. FSL is written mainly by members of the Analysis Group, FMRIB, Oxford, UK. FSL runs on Apple, PCs (Linux and Windows) and Sun, and is very easy to install. Most of the tools can be run both from the command line and as GUIs ("point-and-click" graphical user interfaces).
Referencing
To quote the relevant references for FSL tools you should look in the individual tool's manual page (or the Analysis Group publications page), and also the FSL overview paper:
S.M. Smith, M. Jenkinson, M.W. Woolrich, C.F. Beckmann, T.E.J. Behrens, H. Johansen-Berg, P.R. Bannister, M. De Luca, I. Drobnjak, D.E. Flitney, R. Niazy, J. Saunders, J. Vickers, Y. Zhang, N. De Stefano, J.M. Brady, and P.M. Matthews. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage, 23(S1):208-219, 2004
Next - see the list of tools in FSL.
Introduction
FSL is a comprehensive library of analysis tools for FMRI, MRI and DTI brain imaging data. FSL is written mainly by members of the Analysis Group, FMRIB, Oxford, UK. FSL runs on Apple, PCs (Linux and Windows) and Sun, and is very easy to install. Most of the tools can be run both from the command line and as GUIs ("point-and-click" graphical user interfaces).
Referencing
To quote the relevant references for FSL tools you should look in the individual tool's manual page (or the Analysis Group publications page), and also the FSL overview paper:
S.M. Smith, M. Jenkinson, M.W. Woolrich, C.F. Beckmann, T.E.J. Behrens, H. Johansen-Berg, P.R. Bannister, M. De Luca, I. Drobnjak, D.E. Flitney, R. Niazy, J. Saunders, J. Vickers, Y. Zhang, N. De Stefano, J.M. Brady, and P.M. Matthews. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage, 23(S1):208-219, 2004
Next - see the list of tools in FSL.
Tuesday, May 27, 2008
ParaSolid, geometric modeling kernel, modeleur géométrique
file extension .x_t
There are two main suppliers of kernel software in the market, who provide the basic routines that are used by the overwhelming majority of application software packages. Spatial's ASIC is used to provide the guts for AutoCAD and one or two other packages, while Parasolid, developed by Unigraphics Solutions, is used by most mid-range CAD application developers, such Solid Edge, Solid Works, MicroStation Modeller, Top Solid, etc. Parasolid is also claimed to be the only kernel that is used in high-end 3D modelling systems.
Parasolid is a geometric modeling kernel originally developed by ShapeData, now owned by "Siemens Automation & Drives" former UGS Corp., that can be licensed by other companies for use in their 3D computer graphics software products. It is used in many Computer-aided design (CAD), Computer-aided manufacturing (CAM), Computer-aided engineering (CAE), Product visualization, and CAD data exchange packages, for example: NX (Unigraphics), SolidWorks, SolidEdge, Powershape, T-FLEX CAD, MasterCAM, OneCNC, Virtual Gibbs, DesignFlow, DesignSpace, Renishaw Productivity+, STAR-Design, and Moldflow.
When exported from the parent software package, a Parasolid commonly has the file extension .x_t. Most Parasolid files can communicate and migrate only 3D solids and/or surface data - Parasolid files currently cannot communicate and migrate 2D data such as lines and arcs.
TopSolid est un logiciel de conception assistée par ordinateur conçu et réalisé par Missler Software. Apparu en 1987 sous le nom commercial TopCAD, il était alors l'un des premiers logiciels de CAO volumique fonctionnant sur micro-ordinateur, ce qui lui avait valu la dénomination de "Catia sur micro". Il utilisait alors un modeleur polyédrique c'est-à-dire approximant les formes géométriques par des polyèdres (à facettes planes). TopSolid est donc une CFAO, qui utilise le modeleur géométrique exact ParaSolid, et qui est capable de lire et écrire dans tous les formats ouverts du marché, ainsi que dans quelques formats propriétaires, comme CATIA ou ParaSolid. TopSolid est aujourd'hui diffusé dans le monde entier, et revendique la seconde place française des logiciels de CFAO, derrière Dassault Systèmes avec les logiciels CATIA et Solidworks. Il est même le premier en FAO pure. Il est aussi dans les dix premiers mondiaux.
There are two main suppliers of kernel software in the market, who provide the basic routines that are used by the overwhelming majority of application software packages. Spatial's ASIC is used to provide the guts for AutoCAD and one or two other packages, while Parasolid, developed by Unigraphics Solutions, is used by most mid-range CAD application developers, such Solid Edge, Solid Works, MicroStation Modeller, Top Solid, etc. Parasolid is also claimed to be the only kernel that is used in high-end 3D modelling systems.
Parasolid is a geometric modeling kernel originally developed by ShapeData, now owned by "Siemens Automation & Drives" former UGS Corp., that can be licensed by other companies for use in their 3D computer graphics software products. It is used in many Computer-aided design (CAD), Computer-aided manufacturing (CAM), Computer-aided engineering (CAE), Product visualization, and CAD data exchange packages, for example: NX (Unigraphics), SolidWorks, SolidEdge, Powershape, T-FLEX CAD, MasterCAM, OneCNC, Virtual Gibbs, DesignFlow, DesignSpace, Renishaw Productivity+, STAR-Design, and Moldflow.
When exported from the parent software package, a Parasolid commonly has the file extension .x_t. Most Parasolid files can communicate and migrate only 3D solids and/or surface data - Parasolid files currently cannot communicate and migrate 2D data such as lines and arcs.
Exemple:
TopSolid est un logiciel de conception assistée par ordinateur conçu et réalisé par Missler Software. Apparu en 1987 sous le nom commercial TopCAD, il était alors l'un des premiers logiciels de CAO volumique fonctionnant sur micro-ordinateur, ce qui lui avait valu la dénomination de "Catia sur micro". Il utilisait alors un modeleur polyédrique c'est-à-dire approximant les formes géométriques par des polyèdres (à facettes planes). TopSolid est donc une CFAO, qui utilise le modeleur géométrique exact ParaSolid, et qui est capable de lire et écrire dans tous les formats ouverts du marché, ainsi que dans quelques formats propriétaires, comme CATIA ou ParaSolid. TopSolid est aujourd'hui diffusé dans le monde entier, et revendique la seconde place française des logiciels de CFAO, derrière Dassault Systèmes avec les logiciels CATIA et Solidworks. Il est même le premier en FAO pure. Il est aussi dans les dix premiers mondiaux.
comsol finite element CAD import module
Getting your CAD geometries ready for FEA modeling is easier than ever with the CAD Import Module. It facilitates the reading of industry-standard formats such as STEP, IGES, ACIS® (SAT®) or Parasolid®. Extra add-ons support file formats for packages that have their own geometry kernel.
The CAD Import Module goes beyond just the reading of file formats. The interactive repair feature assures that imported geometries are mathematically correct for FEA modeling. And, in order to cut down on unnecessary details in your CAD geometries, defeaturing tools that remove fillets, small faces, sliver faces, as well as spikes or short edges are included.
The CAD Import Module also provides a bidirectional interface to SolidWorks® that maintains associativity with the CAD system. This means that parameters can be changed in COMSOL models, which result in automatically updating the CAD geometry in SolidWorks that then updates the COMSOL geometry for a new model.
¹ GDS Import requires COMSOL Script.
² Only import of parts is supported.
---
autres formats:
http://www.spinfire-fr.com/SpinFireProfessionnel/FormatsCAODAO/tabid/56/Default.aspx
The CAD Import Module goes beyond just the reading of file formats. The interactive repair feature assures that imported geometries are mathematically correct for FEA modeling. And, in order to cut down on unnecessary details in your CAD geometries, defeaturing tools that remove fillets, small faces, sliver faces, as well as spikes or short edges are included.
The CAD Import Module also provides a bidirectional interface to SolidWorks® that maintains associativity with the CAD system. This means that parameters can be changed in COMSOL models, which result in automatically updating the CAD geometry in SolidWorks that then updates the COMSOL geometry for a new model.
File formats supported by COMSOL products
| Product | File format (file extensions) | Supported versions |
|---|---|---|
| COMSOL Multiphysics | STL (.stl) | |
| VRML (.wrl, .wml) | 1.0 | |
| DXF (.dxf) | up to R14 | |
| GDS (.gds)¹ | 2 | |
| CAD Import Module | Parasolid (.x_t, .x_b) | up to V18 |
| SAT (.sat, .sab) | up to R17 | |
| STEP (.step, .stp) | AP203, AP214 | |
| IGES (.igs, .iges) | up to 5.3 | |
| CATIA V4 Import Module | CATIA V4 (.model) | 4.1.9 to 4.2.4 |
| CATIA V5 Import Module | CATIA V5 (.CATPart, .CATProduct) | R2 to R17 |
| Inventor Import Module | Autodesk Inventor (.ipt)² | 6 to 11 |
| Pro/E Import Module | Pro/Engineer (.prt, .asm) | 16 to Wildfire 3 |
| VDA-FS Import Module | VDA-FS (.vda) | 1 and 2 |
¹ GDS Import requires COMSOL Script.
² Only import of parts is supported.
---
autres formats:
http://www.spinfire-fr.com/SpinFireProfessionnel/FormatsCAODAO/tabid/56/Default.aspx
Sunday, May 25, 2008
Spartan Mac OSX intel
Wavefunction is pleased to announce the release of Spartan'06 for Macintosh – Molecular Modeling for Intel-based Mac's with support for OS X 10.5 (Leopard). This new suite of software is designed to provide Macintosh users with easy to use modeling tools across a wide range of computational chemistry tasks, including conformational searching, calculation of structure, energies, and properties, and quantifying 3-D molecular similarity.
http://www.wavefun.com/products/macintosh/Spartan06/mac_spartan_pricing.html
Spartan'06 is the release of Wavefunction's flagship Spartan line. In addition to the performance, stability, and functionality provided by more than 15 years of professional software development, the following New Features have been added.
---------
chapter 15
This chapter describes functions available under the Setup menu.
Calculations is used to specify molecular mechanics calculations,
semi-empirical calculations, Hartree-Fock molecular orbital
calculations, and correlated calculations, including local density
calculations, density functional calculations, Møller-Plesset
calculations, coupled cluster calculations and quadratic configuration
interaction calculations for ground-state species, and configuration
interaction calculations, local density calculations and (time
dependent) density functional calculations for excited-state species.
Tasks include calculation of energy, equilibrium structure and
conformation, transition-state structure and constructing energy
profiles, although not all tasks are available for some methods. A
wide variety of all-electron Gaussian basis sets are supported for
Hartree-Fock and correlated calculations as are pseudopotentials
for calculations on molecules incorporating heavy elements. Also
provided are a number of thermochemical recipes, including G3 and
G3(MP2), as well as a new (and more economical) parameterized
scheme. Calculations also requests IR, NMR and UV/visible spectra,
and calculation and printing of a variety of molecular properties.
Finally, Calculations identifies libraries and specifies conditions for
identifying similar molecules, based either on molecular structure or
chemical functionality, as well as for identifying molecules that are
compatible with a pharmacophore.
----------
Spartan'06 can retrieve and plot experimental IR (~ 14,000 molecules) and UV/Vis (~ 1,500) spectra from the NIST Chemistry Webbook.
---------
Vertical excitation spectra based using either CIS/CIS(D) or Time Dependent DFT models is provided.
---------
http://www.wavefun.com/products/macintosh/Spartan06/mac_spartan_pricing.html
Spartan'06 is the release of Wavefunction's flagship Spartan line. In addition to the performance, stability, and functionality provided by more than 15 years of professional software development, the following New Features have been added.
---------
chapter 15
This chapter describes functions available under the Setup menu.
Calculations is used to specify molecular mechanics calculations,
semi-empirical calculations, Hartree-Fock molecular orbital
calculations, and correlated calculations, including local density
calculations, density functional calculations, Møller-Plesset
calculations, coupled cluster calculations and quadratic configuration
interaction calculations for ground-state species, and configuration
interaction calculations, local density calculations and (time
dependent) density functional calculations for excited-state species.
Tasks include calculation of energy, equilibrium structure and
conformation, transition-state structure and constructing energy
profiles, although not all tasks are available for some methods. A
wide variety of all-electron Gaussian basis sets are supported for
Hartree-Fock and correlated calculations as are pseudopotentials
for calculations on molecules incorporating heavy elements. Also
provided are a number of thermochemical recipes, including G3 and
G3(MP2), as well as a new (and more economical) parameterized
scheme. Calculations also requests IR, NMR and UV/visible spectra,
and calculation and printing of a variety of molecular properties.
Finally, Calculations identifies libraries and specifies conditions for
identifying similar molecules, based either on molecular structure or
chemical functionality, as well as for identifying molecules that are
compatible with a pharmacophore.
----------
Spartan'06 can retrieve and plot experimental IR (~ 14,000 molecules) and UV/Vis (~ 1,500) spectra from the NIST Chemistry Webbook.
---------
Vertical excitation spectra based using either CIS/CIS(D) or Time Dependent DFT models is provided.
---------
Click here for itemized New Feature List (pdf file)
| Graphical User Interface Features: (RED items not available in Spartan'06 Essential) | |
| Substituent Builder for construction of substituted molecules / virtual libraries | |
| Assignment of Chemical Function Descriptors | |
| Experimental IR and UV/vis spectra available from NIST (on-line) database | |
Molpro quantum chemistry package
Molpro is a complete system of ab initio programs for molecular electronic structure calculations, designed and maintained by H.-J. Werner and P. J. Knowles, and containing contributions from a number of other authors. As distinct from other commonly used quantum chemistry packages, the emphasis is on highly accurate computations, with extensive treatment of the electron correlation problem through the multiconfiguration-reference CI, coupled cluster and associated methods. Using recently developed integral-direct local electron correlation methods, which significantly reduce the increase of the computational cost with molecular size, accurate ab initio calculations can be performed for much larger molecules than with most other programs.
----------
The timings for the standard Molpro benchmark suite:
MOLPRO is not tested regularly on the following machines, but has worked on them in the past..
uv-vis prediction
----------
Molpro benchmarks
The timings for the standard Molpro benchmark suite:
Version 2006.3
Molpro: Secondary platforms
MOLPRO is not tested regularly on the following machines, but has worked on them in the past..
| Machine name | Hardware and operating system | Integer bytes | Other information | Known problems |
| apple-intel | i386-apple-darwin | 4 | Intel Fortran 9.0 |
Computational Methods and Tools
http://www.chem.ucsb.edu/~kalju/comp_tools.html
A good part of gas phase organic and inorganic chemistry can be described well using standard methods of quantum chemistry and statistical mechanics. There are two fundamental reasons why quantum chemistry and statistical mechanics are so successful in describing the chemistry and thermodynamics of molecules in the gas phase chemistry. First, the molecules of interest to the gas phase chemistry are typically small, and thus amenable to highly accurate quantum chemical calculations. Second, isolated molecules or molecular complex in the gas phase have only a few energetically accessible states, and statistical averaging over these states is often feasible. The situation is very different in biochemistry, where one typically studies heterogeneous polymers, such as proteins or nucleic acids, that interact with other molecules in aqueous environment via multitude of weak interactions. Such molecules and molecular complexes are often too large for quantitative description by even the crudest quantum chemical methods, and possess so many energetically accessible states that their complete enumeration is prohibitive. It is clear that research problems in computational biochemistry cannot be solved by simply applying "black box" techniques that have been so successful in characterizing the gas phase chemistry.
 One powerful research strategy to make computational analysis of biomacromolecules feasible involves the combination of reductionism and systems approach. In the first, reductionist stage, a necessary description of components of a biological macromolecule is obtained using quantum mechanics. Then, during the systems approach stage, molecular simulation techniques based on a simplified Hamiltonian are used to describe the structure, motions, and thermodynamics of the whole system. For example, quantum chemistry allows determination of charges, dispersion coefficients (van der Waals parameters), and barriers for torsional motion (flexibility) of components of macromolecules. Quantum chemistry also provides information about the structure, reactivity, and spectroscopic properties of biological molecules. Such information may be needed as input when applying molecular simulation techniques. In summary, quantum chemistry provides an excellent starting point in understanding many biochemical phenomena.
One powerful research strategy to make computational analysis of biomacromolecules feasible involves the combination of reductionism and systems approach. In the first, reductionist stage, a necessary description of components of a biological macromolecule is obtained using quantum mechanics. Then, during the systems approach stage, molecular simulation techniques based on a simplified Hamiltonian are used to describe the structure, motions, and thermodynamics of the whole system. For example, quantum chemistry allows determination of charges, dispersion coefficients (van der Waals parameters), and barriers for torsional motion (flexibility) of components of macromolecules. Quantum chemistry also provides information about the structure, reactivity, and spectroscopic properties of biological molecules. Such information may be needed as input when applying molecular simulation techniques. In summary, quantum chemistry provides an excellent starting point in understanding many biochemical phenomena.
A large number of powerful quantum chemistry programs are available. Below is a list of some quantum chemistry programs that I find useful in my research:
Description of the dynamic behavior and thermodynamic properties of biological macromolecules molecules in solution requires application of molecular simulation techniques. Simulation techniques such as molecular dynamics and Monte Carlo sampling are well established for description of physical properties of fluids. Molecular dynamics simulations have been employed also to describe dynamical structure of proteins, nucleic acids, and their complexes. Free energy simulations provide a theoretically rigorous way to evaluate binding free energies, opening up possibilities to predict efficacy and safety of potential drug candidates.
The field of molecular simulations is plagued by its own problems. For example, it is unlikely that molecular dynamics simulations with currently accessible timescales in the order of 10-100 ns can capture the full dynamics of biological macromolecules. Similarly, it is not clear if currently available molecular mechanics force fields and sampling methods are sufficient for reliable description of binding affinities in novel systems.
Despite these problems, molecular simulations have been proven valuable in understanding chemical reactivity in condensed media and appear promising in understanding the mechanism of enzyme action. Some of the molecular simulation programs that I have used are:

Biological macromolecules are a lot more complex than typical organic molecules. The complexity arises from the large number of atoms in a biological macromolecule, and from the possibility of relatively free rotation around many covalent bonds in a macromolecule. The low rotational barriers give macromolecules flexibility and high conformational complexity. The number of theoretically possible three-dimensional structures that any macromolecule can take is enormous. However, each biological macromolecule adopts a distinct three-dimensional conformation, called the native conformation. Prediction of the native conformation of a unique biomacromolecule based on its covalent structure is a challenging problem, and most information about macromolecular structures is obtained from analysis of X-ray diffraction data from single crystals or from analysis on NMR data of dissolved macromolecules. Further analysis of these structures can be performed using molecular simulation techniques. For example, molecular dynamics simulations allow studying enzyme-substrate complexes, which cannot be studied experimentally due to rapid catalytic turnover under experimental conditions. Visualization tools are indispensable for analysis and presentation of macromolecular structures. Some that I find most useful in my research are:
Computational Biochemistry is Not a Set of Black Box Techniques
A good part of gas phase organic and inorganic chemistry can be described well using standard methods of quantum chemistry and statistical mechanics. There are two fundamental reasons why quantum chemistry and statistical mechanics are so successful in describing the chemistry and thermodynamics of molecules in the gas phase chemistry. First, the molecules of interest to the gas phase chemistry are typically small, and thus amenable to highly accurate quantum chemical calculations. Second, isolated molecules or molecular complex in the gas phase have only a few energetically accessible states, and statistical averaging over these states is often feasible. The situation is very different in biochemistry, where one typically studies heterogeneous polymers, such as proteins or nucleic acids, that interact with other molecules in aqueous environment via multitude of weak interactions. Such molecules and molecular complexes are often too large for quantitative description by even the crudest quantum chemical methods, and possess so many energetically accessible states that their complete enumeration is prohibitive. It is clear that research problems in computational biochemistry cannot be solved by simply applying "black box" techniques that have been so successful in characterizing the gas phase chemistry.
Quantum Chemistry of Biomolecules
One powerful research strategy to make computational analysis of biomacromolecules feasible involves the combination of reductionism and systems approach. In the first, reductionist stage, a necessary description of components of a biological macromolecule is obtained using quantum mechanics. Then, during the systems approach stage, molecular simulation techniques based on a simplified Hamiltonian are used to describe the structure, motions, and thermodynamics of the whole system. For example, quantum chemistry allows determination of charges, dispersion coefficients (van der Waals parameters), and barriers for torsional motion (flexibility) of components of macromolecules. Quantum chemistry also provides information about the structure, reactivity, and spectroscopic properties of biological molecules. Such information may be needed as input when applying molecular simulation techniques. In summary, quantum chemistry provides an excellent starting point in understanding many biochemical phenomena.A large number of powerful quantum chemistry programs are available. Below is a list of some quantum chemistry programs that I find useful in my research:
- Dalton
- Molecular polarizabilities
- Dispersion coefficients (van der Waals parameters)
- DIRCCR12
- Explicitly correlated calculations
- Highly accurate energies
- Gaussian
- All the usual: geometries, isotope effects, conformational search, reaction paths, solvation
- Trickier things: QM/MM via ONIOM, NMR properties, UV-vis
- Gamess
- Environment description via effective fragment potentials
- Renormalized coupled cluster energies
- Jaguar
- Conformational analysis
- Solvation free energies
- SAPT
- Intermolecular forces
- Dispersion interactions
- Molpro
- Multireference systems
- Prediction of UV-Vis spectra
- Spartan
- Electronic structures, conformational analysis
- Visualization of molecular properties
Molecular Simulations
Description of the dynamic behavior and thermodynamic properties of biological macromolecules molecules in solution requires application of molecular simulation techniques. Simulation techniques such as molecular dynamics and Monte Carlo sampling are well established for description of physical properties of fluids. Molecular dynamics simulations have been employed also to describe dynamical structure of proteins, nucleic acids, and their complexes. Free energy simulations provide a theoretically rigorous way to evaluate binding free energies, opening up possibilities to predict efficacy and safety of potential drug candidates.
The field of molecular simulations is plagued by its own problems. For example, it is unlikely that molecular dynamics simulations with currently accessible timescales in the order of 10-100 ns can capture the full dynamics of biological macromolecules. Similarly, it is not clear if currently available molecular mechanics force fields and sampling methods are sufficient for reliable description of binding affinities in novel systems.
Despite these problems, molecular simulations have been proven valuable in understanding chemical reactivity in condensed media and appear promising in understanding the mechanism of enzyme action. Some of the molecular simulation programs that I have used are:
- BOSS
- Parameterization of OPLS-AA force field and conformational analysis
- Monte Carlo simulations of pure liquids, calculation of free energies of solvation
- CHARMM
- Protein structure and dynamics
- Correlation of atomic motions in enzymes
- MCPro
- Monte Carlo simulations of proteins
- Free energies of binding of drug candidates
- NAMD
- Protein structure and dynamics
- Conformational free energies
- TINKER
- Reaction free energies via thermodynamic cycles
- Parameterization of polarizable force fields
Molecular Visualization
Biological macromolecules are a lot more complex than typical organic molecules. The complexity arises from the large number of atoms in a biological macromolecule, and from the possibility of relatively free rotation around many covalent bonds in a macromolecule. The low rotational barriers give macromolecules flexibility and high conformational complexity. The number of theoretically possible three-dimensional structures that any macromolecule can take is enormous. However, each biological macromolecule adopts a distinct three-dimensional conformation, called the native conformation. Prediction of the native conformation of a unique biomacromolecule based on its covalent structure is a challenging problem, and most information about macromolecular structures is obtained from analysis of X-ray diffraction data from single crystals or from analysis on NMR data of dissolved macromolecules. Further analysis of these structures can be performed using molecular simulation techniques. For example, molecular dynamics simulations allow studying enzyme-substrate complexes, which cannot be studied experimentally due to rapid catalytic turnover under experimental conditions. Visualization tools are indispensable for analysis and presentation of macromolecular structures. Some that I find most useful in my research are:
- PyMol
- Raytraced publication-quality images and movies
- Complex scenes thanks to a powerful command language
- gOpenMol
- Analysis of molecular dynamics trajectories
- Visualization and analysis of macromolecular structures
- MOLDEN
- Creation and editing of coordinate files
- Analysis of outputs from quantum chemistry programs
- VMD
- Analysis of molecular dynamics trajectories
- Interactive molecular dynamics
- MOLEKEL
- Visualization and analysis of macromolecular structures
- Visualization of molecular properties from quantum chemistry programs
matlab mac os -or octave
Installation of matlab on linux or macOSX is not so easy.
First the same 3disks but it is different: Mac OS X does not have a Linux kernel and its Darwin kernel is based on FreeBSD and Mach technologies.
Matlab linux macOSX R14 after the name has changed 2007a, 2008 etc...
remarks:
CD2 and CD3 content ureadable data
It works well with mount -o loop .iso /mnt/
For a open source alternative to this, you should check out "octave". It is mostly compatible with matlab.
http://www.gnu.org/software/octave/
i
see http://linuxexpert.wordpress.com/2007/06/18/how-to-install-matlab-7-r14-for-linux/
you need your disks
it’s 3 cd’s collection as ISO images with totally 1.11 GB.
You need 3 files:
-----------
license.dat
license.lic
readme_lic.txt
------------
1. you have to copy license.dat to the installation folder; ~/$MATLAB/.
2. you have to initiate this command from the $MATLAB not from CD.
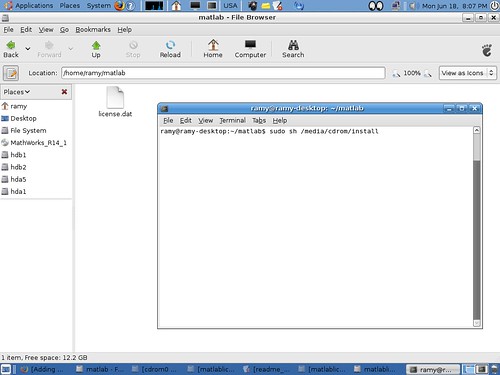
if you are tired from writing cp command for copying files u can initiate the file manager in the root permissions, i do it for ubuntu with Gnome file manager “Nautilus”.
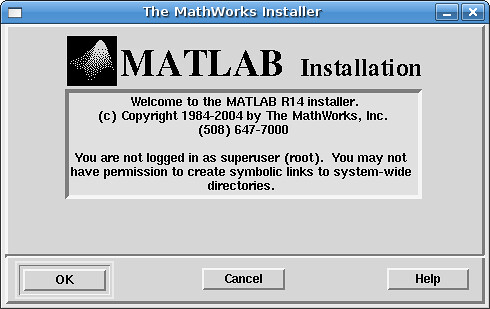
3. Welcome Screen as usual, press OK.
4. License Agreement, press OK.
6. I’m not sure about this message, so proceed and press OK.
7. Also continue, by pressing OK.
8. I’m already chosen all the packages and toolboxes with matlab, press OK.
9. press the button beside “Create symbolic…”, then press OK.
10. press OK.
11. You are asked to put the two other CD’s, press OK.
12. Installation Complete, but the story does not complete :P.
13. You have to open the “license.lic” file and replace the string “your_host_name” with the hostname of the computer where Matlab is being installed, ok if u don’t know what’s the host name go System->Administration->System Monitor-> System tab. In my system it’s ramy-desktop.
14.Copy the edited “license.lic” file to “$MATLAB/etc/”.
15. copy the matlab script file from $MATLAB/bin/scripts to $MATLAB/bin.
16. in the Matlab folder you have install_matlab shell script file, you have to initiate that script you can use this command.
then , u will have a wizard in the terminal, ok it’s sth long, if you are not interested in any modification by accepting the default configurations. i’m a big YES MAN so accepting all of them. if you forget this last step u will have only the matlab running without any functionality because this step makes the required default path configuration and library path configuration.
First the same 3disks but it is different: Mac OS X does not have a Linux kernel and its Darwin kernel is based on FreeBSD and Mach technologies.
Matlab linux macOSX R14 after the name has changed 2007a, 2008 etc...
remarks:
CD2 and CD3 content ureadable data
It works well with mount -o loop .iso /mnt/
For a open source alternative to this, you should check out "octave". It is mostly compatible with matlab.
http://www.gnu.org/software/octave/
i
Starting Installation:
see http://linuxexpert.wordpress.com/2007/06/18/how-to-install-matlab-7-r14-for-linux/
you need your disks
it’s 3 cd’s collection as ISO images with totally 1.11 GB.
You need 3 files:
-----------
license.dat
license.lic
readme_lic.txt
------------
1. you have to copy license.dat to the installation folder; ~/$MATLAB/.
2. you have to initiate this command from the $MATLAB not from CD.
sudo sh /media/cdrom/installif you are tired from writing cp command for copying files u can initiate the file manager in the root permissions, i do it for ubuntu with Gnome file manager “Nautilus”.
sudo cp /media/ramy/license.dat /media/ramy/matlab/etc/
# you can do it in windows-like way
sudo nautilus
3. Welcome Screen as usual, press OK.
4. License Agreement, press OK.
5. u can note that installation directory is the same that you initiate the installer from by the command in step 2, you have to put the license.dat there because the installer test if it’s there then it moves it to ~/$MATLAB/etc. press OK.
6. I’m not sure about this message, so proceed and press OK.
7. Also continue, by pressing OK.
8. I’m already chosen all the packages and toolboxes with matlab, press OK.
9. press the button beside “Create symbolic…”, then press OK.
10. press OK.
11. You are asked to put the two other CD’s, press OK.
12. Installation Complete, but the story does not complete :P.
13. You have to open the “license.lic” file and replace the string “your_host_name” with the hostname of the computer where Matlab is being installed, ok if u don’t know what’s the host name go System->Administration->System Monitor-> System tab. In my system it’s ramy-desktop.
14.Copy the edited “license.lic” file to “$MATLAB/etc/”.
15. copy the matlab script file from $MATLAB/bin/scripts to $MATLAB/bin.
16. in the Matlab folder you have install_matlab shell script file, you have to initiate that script you can use this command.
sudo sh install_matlabthen , u will have a wizard in the terminal, ok it’s sth long, if you are not interested in any modification by accepting the default configurations. i’m a big YES MAN so accepting all of them. if you forget this last step u will have only the matlab running without any functionality because this step makes the required default path configuration and library path configuration.
Subscribe to:
Posts (Atom)