
The potential functions representing the non-bonded energy are formulated as a sum over interactions between the particles of the system.
The simplest choice, employed in many popular force field (physics), is the "pair potential", in which the total potential energy can be calculated from the sum of energy contributions between pairs of atoms. An example of such a pair potential is the non-bonded Lennard–Jones potential (also known as the 6–12 potential), used for calculating van der Waals forces.
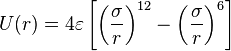
Another example is the Born (ionic) model of the ionic lattice. The first term in the next equation is Coulomb's law for a pair of ions, the second term is the short-range repulsion explained by Pauli's exclusion principle and the final term is the dispersion interaction term. Usually, a simulation only includes the dipolar term, although sometimes the quadrupolar term is included as well.(Usually known as Buckingham potential model)
Bond order potential is a class of empirical (analytical) interatomic potentials which is used in molecular dynamics and molecular statics simulations. Examples include the Tersoff potential, the Brenner potential, the Finnis-Sinclair potentials, ReaxFF, and the second-moment tight-binding potentials.
They have the advantage over conventional molecular mechanics force fields in that they can, with the same parameters, describe several different bonding states of an atom, and thus to some extent may be able to describe chemical reactions correctly. The potentials were developed partly independently of each other, but share the common idea that the strength of a chemical bond depends on the bonding environment, including the number of bonds and possibly also angles and bond length. It is based on the Linus Pauling bond order concept and can be written in the form
This means that the potential is written as a simple pair potential depending on the distance between two atoms r_{ij}, but the strength of this bond is modified by the environment of the atom i via the b_{ijk} term. Alternatively, the energy can be written in the form

where 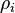 is the electron density at the location of atom
is the electron density at the location of atom 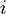 . These two forms for the energy can be shown to be equivalent.
. These two forms for the energy can be shown to be equivalent.
is the electron density at the location of atom . These two forms for the energy can be shown to be equivalent.
The Tersoff potential (which was originally used to simulate carbon, silicon and germanium and has since been used for a wide range of other materials) involves a sum over groups of three atoms, with the angles between the atoms being an important factor in the potential.
Other examples are the embedded-atom method (EAM) and the Tight-Binding Second Moment Approximation (TBSMA) potentials, where the electron density of states in the region of an atom is calculated from a sum of contributions from surrounding atoms, and the potential energy contribution is then a function of this sum.
------------------
Most classical force fields implicitly include the effect of polarizability, e.g. by scaling up the partial charges obtained from quantum chemical calculations. These partial charges are stationary with respect to the mass of the atom. But molecular dynamics simulations can explicitly model polarizability with the introduction of induced dipoles through different methods, such as Drude particles or fluctuating charges. This allows for a dynamic redistribution of charge between atoms which responds to the local chemical environment.
For many years, polarizable MD simulations have been touted as the next generation. For homogenous liquids such as water, increased accuracy has been achieved through the inclusion of polarizability.
Some promising results have also been achieved for proteins. However, it is still uncertain how to best approximate polarizability in a simulation.
Thanks for the best content
ReplyDeleteGREAT WORK
IMPRESSIVE!!!
REALLY APPRECIATE YOUR WORK!!!
VIRTUAL SERVICES INDIA
VIRTUAL SERVICES UK